
What is Achondroplasia
Achondroplasia is a skeletal dysplasia (dysplasia - abnormal growth or development), also identified as a rare bone disease.
For starters, the term rare disease identifies a condition or a syndrome or a disorder that is very uncommon, affecting less that 1 person in 2000, in the european designation [1, 2]. In the american designation, however, this definition is a little narrower: a disease is considered rare in the US if it affects fewer than 200 000 people at any given time [3]. Using the current estimate of total population in the US, about 325 million people, according to the US Census, it's about 1 in 1,600 (but since it's based on prevalence on the population, the ratio will change with changes in total population) [4].
In Europe, a rare disease may affect from only a few patients to nearly 250 000, depending on the rarity of the condition. There are more than 30 million people affected by rare diseases in Europe of which there are about 6 800 in existance [2].
80% of rare diseases have identified genetic origins whilst others are the result of infections (bacterial or viral), allergies and environmental causes or are degenerative and proliferative [3].
50-75% of rare diseases start in childhood [2].
Achondroplasia is the most common form of chondrodysplasia and occurs due to mutations in the fibroblast growth factor receptor 3 (FGFR3) gene, encoding a transmembrane receptor (a protein that spans the membrane of cells) that is important in regulating linear bone growth, among other functions [5, 6]. Diagnosis is based on the presence of characteristic clinical and radiological findings, and so, prenatal diagnosis can occur incidentally during routine prenatal ultrasound examination in the 3rd trimester and can be confirmed by amniocentesis by finding the common FGFR3 mutation [7, 8]. Without genetic testing, misdiagnosis can occur, and so, incorrect counseling may be given to parents [9]. Pre-implantation genetic diagnosis is possible in specialized laboratories.
|
Mutation: permanent alteration in the DNA, in a specific point of a gene or in more than one. It can either have no effect, alter the product of a gene, or prevent the gene from functioning properly or completely.
|
Although this condition is autossomal dominant, meaning that if only one parent suffers from achondroplasia there's a 50% chance of passing it on to their offspring, 80% of cases are due to a de novo (new) mutation in children from parents with average height. Carriers of the disease can only be hetrozygous, since homozygous achondroplasia is lethal and genetic counseling is advised.
It is estimated that its incidence is about 1/25,000 live births worldwide and some of the characteristic clinical features, shown below, are visible at birth [8]:
Rhizomelia – disproportion of the length of the proximal limb.
Rhizomelia – disproportion of the length of the proximal limb.

| Terminology used to describe every part of the body unambiguously. Credits: Anatomic terms of location -Wikipedia |
Exaggerated lumbar lordosis – Excessive curvature of the lower back.

| Credits: knowhowmd.com |
Brachydactyly – The shortness of the fingers is relative to the length of other long bones and other parts of the body. The hands are also broad, short and trident shaped.
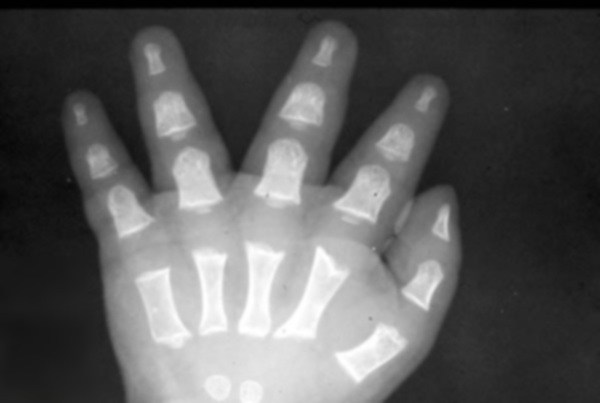
| Credits: Rixir.co.pk |
Macrocephaly and frontal bossing – Is an unusually large head with a prominent forehead.
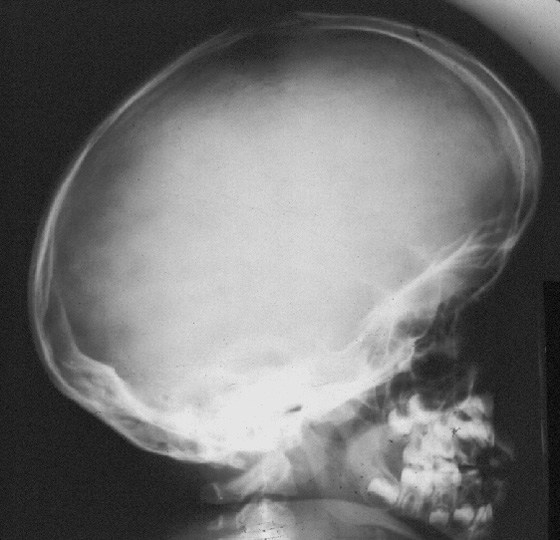
| Credits: Gamuts.isradiology.org |
Midface hypoplasia – The images below showing a normal profile at 28 weeks and a profile at 30 weeks with midface hypoplasia with frontal bossing in a fetus with achondroplasia. In combination with adenoid and tonsil hypertrophy it can lead to obstructive sleep apnoea [10]. Treatment of obstructeive sleep apnea may include adenotonsillectomy, weight loss, and/or continuous positive airway pressure (CPAP).
Midface hypoplasia is also thought to be the cause of speech development delays, shown by about 20% of children with achondroplasia [11]. Speech therapy can be offered if concerns arise.
| Obstructive sleep apnoea: significant or complete, repetitive airflow reduction during sleep due to an obstruction, in this case, the airways themselves, the adenoids and tonsils. |
Midface hypoplasia is also thought to be the cause of speech development delays, shown by about 20% of children with achondroplasia [11]. Speech therapy can be offered if concerns arise.

| Credits: (Ramos et al., 2008). |
Thoracolumbar kyphosis – is very common in children with achondroplasia and it's usually noticed during infancy. It is due to mechanical factors, specifically the general muscular hypotonia which, along with small limbs, small neck and large head, make achievement of gross motor skills (larger movements) slower than typical.

| Original photo. |
Genu varus – Is a physical deformity marked by outward bowing of the lower leg in relation to the thighbow legs, which often occurs in childhood. Surgical correction can re-align bowing of legs.
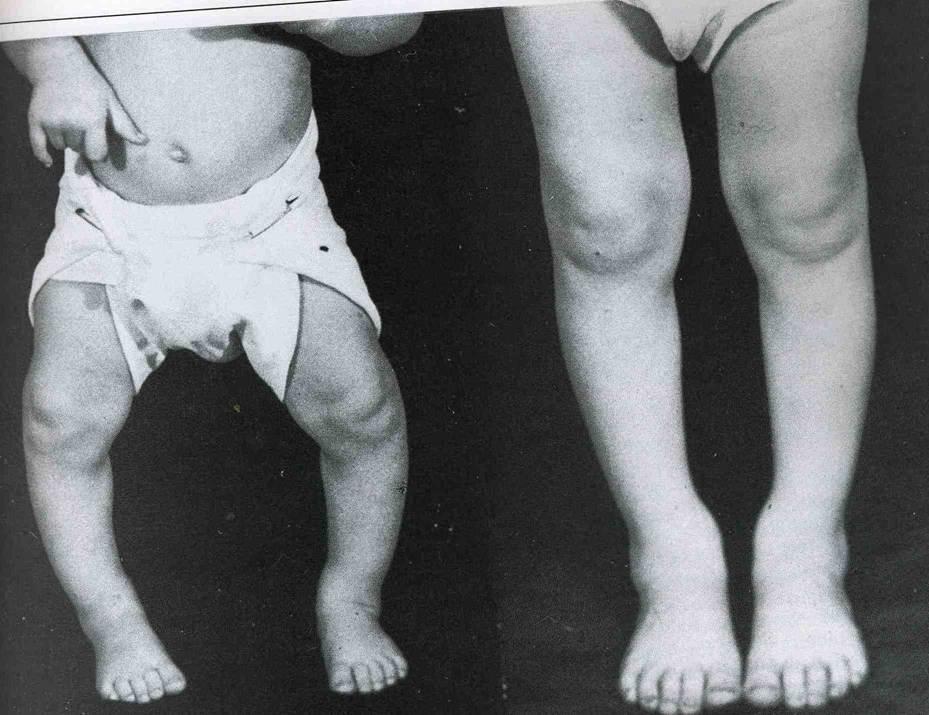
| Credits: Studyblue.com |
Many other complications arise from early childhood, to adulthood, such that management is multidisciplinary and anticipatory care is essential:
Most joints can be hyperextensible.
Otitis media is a common issue, and chronic otitis media is present in 25% of infants with achondroplasia. It is caused by the small proportions of the skull (small Eustachian tubes, also known as auditory tubes, right behind the ear drum, small pharynx and large tonsils and adenoids) and can lead to hearing problems, including conductive hearing loss (problems in the middle ear or ear drum), which is present in about 40% of adults with achondroplasia. Treatment of ear infections and serious otitis media, along with assessment of any hearing problems is needed [8].
Dental crowding is common and removal of some teeth may be necessary for dental alignment [4].
Cord compression at the level of the foramen magnum (shown in the image below) can be encountered in infancy and early childhood causing central sleep apnea, developmental delay, and long-track signs (muscle spasms and involuntary muscle contractions). This problem tends to solve itself with growth since the size of the foramen magnum tends to increase, relative to the size of the spinal cord. Still, surgical decompression of the foramen magnum may be needed in infants. Activities which lead to a risk of injury to the craniocervical junction should be avoided.
While cord compression at the level of the foramen magnum tends to get better with growth, spinal stenosis, and its accompanying neurological deficits (such as neurological claudication), has an increased frequency in adulthood, especially in those with presistent kyphosis. This is due to a relative reduction in spinal canal size when compared to the spinal cord's size and is aggravated by obesity, which is a also a common problem and also leads to cardiovascular complications [5, 12]. Lumbar laminectomy may be necessary in order to treat spinal stenosis and weight must be controlled to avoid complications such as this one [8].
There is also a small risk of hydrocephalus, with raised intracranial venous pressure.
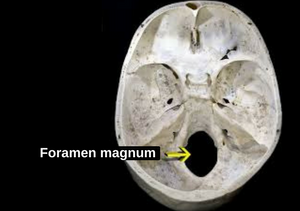
| Foramen Magnum - where the spinal cord connects to the brain. Credits: Foramen magnum - Wikipedia. |
Beside rhizomelia, skeletal X-rays demonstrate generalized metaphyseal irregularities, narrowing of the interpedicular distance of the lower lumbar vertebrae and an abnormal pelvis with small square iliac wings and narrow sacrosciatic notch. Affected women must deliver by caesarian section due to small pelvis size.
Adults reach a height of 131±5.6 cm (men) and 124±5.9 cm (women). Despite all the above complications (which can be seen in more detail here), there is only a slight decrease in life expectancy compared to the general population, potentially due to cardiovascular disease [8].
Sources:
- European Medicines Agency. Orphan designation. [cited 2017 10/05]; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000029.jsp&mid=WC0b01ac05800240ce.
- Bavisetty, S., W.W. Grody, and S. Yazdani, Emergence of pediatric rare diseases: Review of present policies and opportunities for improvement. Rare Diseases, 2013. 1: p. e23579. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3932940/.
- Eurordis. Rare Diseases : Understanding this Public Health Priority. 2005 [cited 2017 10/05]; Available from: http://www.eurordis.org/IMG/pdf/princeps_document-EN.pdf.
- U.S. Department of Commerce. U.S. and World Population Clock. United States Census Bureau 2017; Available from: https://www.census.gov/popclock/.
- Horton, W.A., J.G. Hall, and J.T. Hecht, Achondroplasia. The Lancet, 2007. 370(9582): p. 162-172. Available from: http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(07)61090-3/fulltext(07)61090-3/fulltext.
- Rousseau, F., et al., Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature, 1994. 371(6494): p. 252-4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8078586.
- Proud, V.K. and E.R. Elias, Chapter 26 - GENETIC SYNDROMES AND DYSMORPHOLOGY, in Developmental-Behavioral Pediatrics (Fourth Edition). 2009, W.B. Saunders: Philadelphia. p. 246-257. Available from: http://www.sciencedirect.com/science/article/pii/B9781416033707000262.
- Bober, M. and A. Duker. Achondroplasia. 2013 [cited 2017 10/05]; Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=148&Disease_Disease_Search_diseaseGroup=achondroplasia&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Achondroplasia&title=Achondroplasia&search=Disease_Search_Simple.
- Trotter, T.L. and J.G. Hall, Health Supervision for Children With Achondroplasia. Pediatrics, 2005. 116(3): p. 771-783. Available from: http://pediatrics.aappublications.org/content/pediatrics/116/3/771.full.pdf.
- American Academy of Sleep Medicine, International classification of sleep disorders, revised: Diagnostic and coding manual. 2001: American Academy of Sleep Medicine. Available from: http://www.esst.org/adds/ICSD.pdf.
- Hunter, A.G., et al., Medical complications of achondroplasia: a multicentre patient review. Journal of Medical Genetics, 1998. 35(9): p. 705-712. Available from: http://jmg.bmj.com/content/jmedgenet/35/9/705.full.pdf.
- Pauli, R.M. and C.A.F.D.J. Wilkin, Achondroplasia. 1998 [Updated in 2012], University of Washington, Seattle (WA): GeneReviews. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1152/.

